
Càncer hereditari
Importància i necessitat de detectar-lo

Encara que tan sols l’1% dels casos de càncer es poden considerar de tipus hereditari, en l’actualitat s’han descrit més de 200 síndromes caracteritzades per l’agregació familiar de distints tipus de tumors. La identificació al llarg del segle xx de molts dels gens responsables d’aquestes síndromes ha permès un gran avenç en la caracterització d’aquestes famílies a l’hora d’identificar o descartar els individus en risc i establir mesures preventives específiques. No obstant això, existeix encara un alt percentatge de casos hereditaris en què la causa de la susceptibilitat és desconeguda, la qual cosa fa de la recerca de nous gens mitjançant les noves tecnologies una de les prioritats en investigació.
Paraules clau: càncer hereditari, models d’herència, BRCA1, BRCA2, síndrome de càncer de pit i ovari hereditari.
INTRODUCCIÓ HISTÒRICA
A hores d’ara s’han identificat més de 200 síndromes hereditàries associades amb un increment en el risc de càncer, caracteritzades per l’aparició de neoplàsies malignes primàries en múltiples membres d’una família i que en alguns casos s’associen amb anomalies congènites. Va ser precisament l’observació d’agrupacions familiars d’aquestes anomalies congènites el que va portar a observar també els primers casos de càncer hereditari. Entre aquests primers es troben els casos de neurofibromatosi tipus 1 (NF1), caracteritzats per la presència d’alteracions cutànies, com les «taques cafè amb llet» i nòduls petits en la pell, a més d’un risc incrementat al desenvolupament de tumors del sistema nerviós central.
El 1866, el cirurgià francès Paul Broca va publicar per primera vegada un exemple de càncer de mama hereditari en què descrivia quatre generacions de dones afectades de càncer de mama en la família de la seua dona. Al llarg del segle xx es van realitzar nombroses descripcions clíniques d’agrupacions familiars de càncer, no obstant això van ser la teoria de Knudson a començament dels anys setanta (Knudson, 1971) i el descobriment l’any 1986 del primer gen de susceptibilitat al càncer, Rb, implicat en el retinoblastoma familiar (Friend et al., 1986), les dues fites que van marcar una nova era en el camp del càncer hereditari.

Imatges de la sèrie «David contra Goliat», on el fotògraf Tino Soriano continua reflectint el càncer, en concret l’infantil. En la imatge, Hospital de Sant Pau de Barcelona. / Tino Soriano
La teoria de Knudson es basava en l’observació de 48 casos de retinoblastoma, un tumor maligne de retina. L’any 1971, Knudson va desenvolupar la seua teoria de two-hits, després d’observar que els casos de retinoblastoma hereditari presentaven una edat d’aparició molt més primerenca que els casos esporàdics (casos únics sense antecedents familiars). Segons la hipòtesi de Knudson, la malaltia és causada per dos esdeveniments mutacionals, two-hits, que afecten les dues còpies del gen causant de la malaltia. En la forma hereditària, una mutació (alteració en la informació genètica) es deu trobar en la línia germinal, es transmet de generació en generació i confereix als portadors una susceptibilitat molt alta a patir el càncer. Aquesta susceptibilitat s’heretaria de manera autosòmica dominant, és a dir, un gen anormal d’un dels pares seria prou per causar la susceptibilitat a patir la malaltia, encara que la segona còpia del gen de l’altre progenitor fóra normal. La segona còpia s’alteraria a escala somàtica, en aquest cas la retina, i és llavors que es produiria el tumor (figura 1). En les formes no hereditàries, ambdues mutacions ocorrerien a escala somàtica.
Poc després es va publicar que aquest model pot ser extensible al tumor de Wilms (tumor renal en la infantesa), al neuroblastoma en la infantesa (tumor embrionari del sistema nerviós) i al feocromocitoma (tumor de la glàndula suprarenal) i en general a tots els càncers hereditaris. Aquesta teoria s’ha mantingut fins a l’actualitat i, encara que hi ha excepcions, la majoria de les principals síndromes de càncer hereditari segueixen aquest patró.
PRINCIPALS GENS IMPLICATS: MODELS D’HERÈNCIA
Fins a les acaballes del segle xx, la investigació sobre la susceptibilitat heretada a patir càncer es va centrar en la identificació d’aquest tipus de gens d’alta penetrància o alta susceptibilitat. Exemples clàssics de gens, a més dels ja mencionats, són el gen TP53, identificat l’any 1990 i responsable de la síndrome de Li-Fraumeni; el gen APC, en l’any 1991, i responsable del càncer de còlon polipòsic familiar o el gen MSH2, de 1993, implicat en el càncer de còlon no polipòsic hereditari. L’any 1994 va ser especialment fructífer, ja que es van identificar tres dels gens més importants, MLH1, segon gen implicat en el càncer de còlon no polipòsic, BRCA1, primer gen implicat en la síndrome de càncer de mama i ovari hereditari (CMOH), i p16, implicat en el melanoma familiar. Aquests són alguns dels més importants. Destaca finalment el descobriment el 1995 de BRCA2, el segon gen de susceptibilitat per a càncer de mama i ovari, ja que utilitzarem l’exemple d’aquesta síndrome per a introduir altres conceptes en aquest article. L’origen del càncer resideix en la capacitat que adquireixen certes cèl·lules per a escapar dels mecanismes que regulen el creixement cel·lular normal i que arriben a proliferar de manera descontrolada. Tots els gens mencionats fins ara, i la majoria dels implicats en càncer hereditari, són els denominats gens supressors de tumors (GST). En general la funció d’aquests gens és controlar de manera negativa el cicle cel·lular i impedir aquesta proliferació descontrolada de cèl·lules. Hi ha algunes excepcions a aquesta regla, en les quals el gen implicat no és un GST sinó un protoncogén, és a dir, un gen la funció del qual és l’activació del creixement i la divisió cel·lular. Un exemple d’aquest tipus de gens és el protoncogén RET, implicat en la neoplàsia endocrina múltiple tipus 2 (MEN2). La diferència amb els GST és que el seu paper és activador del cicle cel·lular i que pateixen mutacions activadores de funció, per la qual cosa només és necessari que estiga alterada una còpia del gen perquè es produesca el tumor.
«El 1866, el cirurgià francès Paul Broca va publicar per primera vegada un exemple de càncer de mama hereditari en què descrivia quatre generacions de dones afectades de càncer de mama»
En tots els casos esmentats fins ara, el patró d’herència és autosòmic dominant, no obstant això és obligat mencionar algunes síndromes de càncer hereditari que segueixen un model autosòmic recessiu. En aquests casos, les dues còpies del gen han d’estar mutades a nivell germinal perquè es desenvolupe la malaltia. La majoria dels gens implicats en aquestes síndromes actuen en processos de reparació de l’ADN, la qual cosa és la causa que la inestabilitat cromosòmica hi siga un tret comú. Un exemple molt significatiu és l’anèmia de Fanconi (AF), una malaltia que es caracteritza per fallida medul·lar progressiva, hipersensibilitat a agents intercalants de l’ADN (molècules que s’intercalen en les bases que conformen l’ADN i que n’afecten l’estructura) i susceptibilitat incrementada a patir càncer.
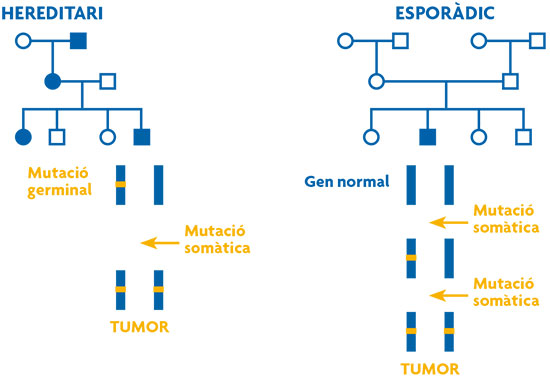
Figura 1. Model de two-hits de Knudson. S’hi presenten dos arbres genealògics en què els cercles representen dones i els quadrats homes. S’hi representen en color blau els individus diagnosticats de càncer. En el cas hereditari (a l’esquerra), una de les còpies del gen responsable de la malaltia ja està alterada en la línia germinal, això fa que l’esdeveniment d’una segona mutació somàtica siga molt probable i per tant el risc de desenvolupament del càncer siga molt alt i l’edat d’aparició més primerenca. En el cas esporàdic (a la dreta) ambdues mutacions han d’ocórrer a nivell somàtic, succés molt més improbable i que explica per què en aquesta família hi ha un sol cas de càncer i l’edat d’aparició és més tardana. / Ana Osorio
GESTIÓ CLÍNICA DE LES FAMÍLIES AMB CÀNCER HEREDITARI
Cal puntualitzar que aquests que podem considerar com clarament hereditaris, globalment tan sols representen l’1% dels casos de càncer, que la major part de les vegades apareixen de manera esporàdica i que en la seua etiologia participen multitud de factors.
No obstant això, encara que beneficia un percentatge petit dels casos, la identificació d’aquests gens d’alt risc ha representat un gran avenç en la gestió de les famílies amb càncer hereditari, ja que ofereix la possibilitat de realitzar un test genètic mitjançant el qual identificar i descartar els individus en risc. Actualment l’única opció que existeix per a curar un pacient de càncer és una detecció primerenca o bé la prevenció. En aquest sentit, la possibilitat d’identificar en una família amb càncer hereditari quina és la causa de la susceptibilitat permet fer un seguiment personalitzat d’aquells membres de la família que són portadors d’una mutació concreta i que sabem positivament que tenen un risc molt alt de desenvolupar un càncer al llarg de la seua vida.
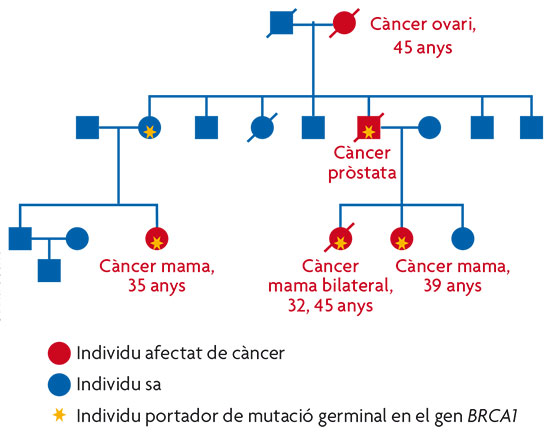
Figura 2. Exemple d’una família amb càncer de mama hereditari, portadora de mutació germinal en el gen BRCA1. S’hi observen característiques que són comunes a la majoria dels càncers familiars, entre altres: un elevat nombre de persones afectades en la família, una edat d’aparició dels tumors més primerenca que l’edat mitjana de diagnòstic del tumor en la població general, presència de bilateralitat en el cas que el càncer afecte òrgans parells i presència d’altres tumors associats, com en aquest cas, càncer d’ovari. / Ana Osorio
Prenguem com a exemple la família amb càncer de mama i ovari hereditari representada en la figura 2. Una vegada detectada la mutació en BRCA1 responsable de la susceptibilitat, és possible realitzar el test genètic en totes aquelles dones de la família que no han desenvolupat càncer. Les que resulten portadores, i per tant tinguen un risc elevat, podran ser sotmeses a programes de seguiment personalitzats que permetran detectar el tumor en una fase molt primerenca en la qual serà curable amb una alta probabilitat. En aquestes dones en risc, depenent de les circumstàncies personals, existeix fins i tot la possibilitat de realitzar una cirurgia profilàctica que reduesca la possibilitat de patir càncer de mama o ovari fins en un 95%. Depenent del tipus de tumor hereditari, la detecció de l’alteració responsable de la susceptibilitat permet prendre mesures més o menys efectives.
Per exemple, la síndrome de MEN2A es caracteritza per l’aparició de tumors endocrins, en particular carcinoma medul·lar de tiroide, feocromocitoma i hiperplàsia de paratiroides, i és causada en la majoria dels casos per mutacions heretades en el protoncogén RET mencionat prèviament en aquest article. La naturalesa d’aquest gen fa que els individus portadors de mutacions desenvolupen un tumor en el 100% dels casos, ja que només cal que una còpia del gen estiga alterada, alteració que representa una excepció al model de two-hits de Knudson. La mesura principal d’intervenció preventiva primària és una tiroïdectomia profilàctica en edat infantil que elimina per complet el risc de desenvolupar carcinoma medul·lar de tiroide.
En l’extrem oposat es troben les síndromes associades amb l’aparició de múltiples tumors, com el de Li-Fraumeni, caracteritzat per l’aparició de sarcomes de parts toves, osteosarcomes, càncer de mama, tumors cerebrals i leucèmia. En aquesta síndrome el seguiment dels pacients és molt més complicat, no sols per la possibilitat de desenvolupar tumors distints sinó perquè n’hi ha que són molt difícils de diagnosticar precoçment.
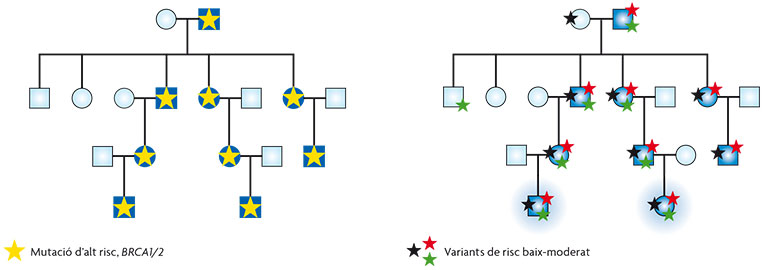
Figura 3. L’arbre genealògic de l’esquerra representa un cas clàssic de càncer hereditari en què tots els individus afectats de la família (en blau) s’expliquen per una mutació en un gen d’alt risc (estrella groga). La genealogia de la dreta representa la mateixa família, però aquesta vegada, els casos de càncer s’expliquen per l’acumulació i combinació de variants de baix i/o moderat risc. Només els casos en què s’acumula un nombre suficient de variants arribaran a desenvolupar càncer, mentre que els individus que presenten una sola variant no el desenvoluparan, ja que el risc que aquestes confereixen és molt baix a nivell individual. / Ana Osorio
ALTRES GENS DE SUSCEPTIBILITAT
Un dels principals problemes que es presenten quan ens enfrontem a un cas de càncer hereditari és la no detecció de l’alteració causant de la susceptibilitat. En la majoria de les síndromes de càncer hereditari, hi ha un percentatge dels casos que no s’expliquen per mutacions en els gens coneguts, un dels exemples més representatius d’aquest problema és la síndrome de CMOH.
«L’origen del càncer resideix en la capacitat que adquireixen certes cèl·lules per a escapar dels mecanismes que regulen el creixement cel·lular normal i que arriben a proliferar de manera descontrolada»
Quan es van identificar els gens BRCA1 i BRCA2 fa quasi vint anys, es va pensar que les mutacions germinals en aquests podrien explicar fins al 80% dels casos de càncer de mama hereditari, no obstant això pocs anys després es va confirmar que aquest percentatge no era tan alt i que podia ser variable segons la població analitzada. Actualment es considera que en general, aquests dos gens no expliquen més d’un 20% dels casos, la qual cosa ha motivat que en els darrers deu anys la recerca de nous gens de susceptibilitat haja estat un dels principals objectius dels grups d’investigació. No obstant això, des dels anys noranta no s’ha identificat cap altre gen d’alt risc específicament implicat en la síndrome. Això ha portat a l’acceptació del model poligènic, que explicaria l’increment en el risc i l’agrupació familiar, per la combinació de variants de risc baix o moderat que s’acumularien en aquestes famílies (figura 3).
A escala individual, cada variant modificaria molt poc el risc, però en unió amb altres factors, tant de genètics com d’ambientals, aquests gens de baixa o moderada penetrància no sols estarien implicats en aquests patrons familiars, sinó que serien responsables de les diferències en la susceptibilitat a patir càncer que hi ha entre els individus de la població general. En els últims cinc anys s’ha produït un gran avenç en el descobriment d’algunes d’aquestes variants gràcies als estudis d’associació de genoma complet, coneguts comunament com GWAS (Genome Wide Association Studies) (Visscher, 2012). Aquest model s’està aplicant també a altres tipus de càncer hereditari.
Bibliografia
Cirulli, E. T. i D. B. Goldstein, 2010. «Uncovering the Roles of Rare Variants in Common Disease Through Whole-genome Sequencing». Nature Reviews Genetics, 11: 415-425. DOI: <10.1038/nrg2779>.
Friend, S. H. et al., 1986. «A Human DNA Segment with Properties of the Gene that Predisposes to Retinoblastoma and Osteosarcoma». Nature, 323: 643-646. DOI: <10.1038/323643a0>.
Knudson Jr., A. G., 1971. «Mutation and Cancer: Statistical Study of Retinoblastoma». PNAS, Proceedings of the National Academy of Sciences, 68(4): 820-823. DOI: <10.1073/pnas.68.4.820>.
Ponder, B. A.; Antoniou, A.; Dunning, A.; Easton, D. F. i P. D. Pharoah, 2005. «Polygenic Inherited Predisposition to Breast Cancer». Cold Spring Harbor Symposia Quantitative Biology, 70: 35-41. DOI: <10.1101/sqb.2005.70.029>.
Visscher, P. M.; Brown, M. A.; McCarthy, M. I. i J. Yang, 2012. «Five Years of GWAS Discovery». The American Journal of Human Genetics, 90: 7-24. DOI: <10.1016/j.ajhg.2011.11.029>.




