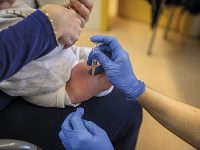

Què es commemora l’últim dia de febrer? Què és una malaltia minoritària? L’última jornada de febrer és el Dia Mundial de les Malalties Rares, un conjunt de trastorns que són difícils de visibilitzar per la poca prevalença que presenten. Aquesta data va ser establerta l’any 2008 (fent-la coincidir amb un 29 de febrer, precisament, per ser un dia «estrany») per l’Organització Europea de Malalties Rares, EURORDIS, motivada per l’insuficient tractament i atenció que reben les persones amb malalties minoritàries i les seves famílies.
Com afirma la investigadora i neuropediatra de l’hospital Sant Joan de Déu Mercedes Serrano en La sociedad civil y las enfermedades raras, no existeix una definició única i universalment acceptada de malaltia rara: per a la Unió Europea són les que afecten menys de 5 persones de cada 10.000 (o, cosa que és el mateix, menys d’1 de cada 2.000) mentre que a altres llocs, com els Estats Units, es consideren xifres absolutes (malalties que afecten menys de 200.000 persones).
«A banda del desconeixement, el principal problema de les malalties minoritàries és la falta d’investigació»
L’Organització Mundial de la Salut (OMS) calcula que hi ha 7.000 tipus diferents de malalties rares. Amb aquesta xifra tan alta es dona l’anomenada paradoxa de la raresa: encara que cadascuna d’elles, de forma aïllada, sigui molt poc freqüent, totes juntes impliquen una gran quantitat de població afectada. La Unió Europea estima que el conjunt de malalties minoritàries afecten entre el 6 % i el 8 % de la població, és a dir, entre 27 i 36 milions de persones. Amb aquestes xifres, la Federació Espanyola de Malalties Rares, FEDER, calcula que a Espanya hi ha uns 3 milions de persones afectades.
Almenys el 80 % de les malalties rares tenen un origen genètic, per tant crònic, i per aquest motiu moltes d’elles es manifesten durant l’edat pediàtrica. Tanmateix, tot i que les manifestacions de les malalties solen ser durant l’etapa primària de la vida, no es restringeixen a un període concret. És a dir, poden aparèixer en el naixement, a l’edat adulta o, fins i tot, en la senectut.

A banda del desconeixement, el principal problema de les malalties minoritàries és la falta d’investigació. Això provoca manca d’informació, d’estudis, de tractaments que puguin facilitar la vida quotidiana dels pacients perquè més enllà de xifres i definicions, està la realitat de qui són veritables protagonistes: les persones afectades i les seves famílies.
Síndrome de Williams
Álvaro té dos anys. Va néixer amb una fissura de la mida de l’ungla del dit polze al paladar tou. Quan la seva mare, Áurea, li donava a menjar, la llet sortia cap a l’exterior del nas a través d’aquest «forat». Després de més visites al pediatre de les que li corresponien, l’única afectació diagnosticada era la del paladar. Però amb només un mes i mig de vida, va deixar de menjar un dia sencer. Aquella mateixa nit a urgències descobriren que tenia un buf al cor. En pocs dies i moltes proves, el diagnòstic ja era clar: síndrome de Williams, una alteració genètica produïda per una deleció (pèrdua de part del material genètic) en el cromosoma 7. Això produeix retard en el desenvolupament mental, trets facials molt distintius i un estrenyiment de l’artèria aorta. Afecta 1 de cada 7.500 naixements.
Amb dos anys i mig, Álvaro fa unes setmanes que va començar a caminar. Tampoc parla, només pot pronunciar mamà i papà. Amb només tres mesos la família va començar amb la teràpia i l’atenció precoç. La mare confessa que, en poc temps, el canvi va ser radical: «Al principi allà on el posàvem, es quedava. No era capaç de girar-se ni d’alçar els braços». Durant el primer any de vida no podia agafar un refredat perquè les seves branques pulmonars són extremadament fines. A més, l’havien d’alimentar amb un biberó especial perquè amb la fissura al paladar no podia succionar amb normalitat. Però superats aquests inicis, tot va començar a millorar.
Per a aquesta síndrome no hi ha tractament ni cura. Les intervencions i diferents teràpies que es realitzen són útils per a pal·liar o reduir alguns dels símptomes, però com que es tracta d’una malaltia que afecta parts diferents del cos, ha de ser un equip multidisciplinari qui s’encarregui de les teràpies.
Pel que fa a l’escolarització, en el cas de la síndrome de Williams s’ha produït debat al voltant de la inclusió i sobre si portar els menors afectats a un centre d’educació especial o a un ordinari. Áurea explica que són nens que tendeixen molt a la imitació i que, per tant, necessiten tenir de referència companys que portin a terme les activitats corresponents a la seva edat: «Si el matriculem en un centre d’educació especial, poc aprendrà. El portarem a un col·legi ordinari amb suports especials, perquè l’hem de motivar».
Quan Álvaro va ser diagnosticat amb aquesta malaltia minoritària, a Mallorca (el seu lloc d’origen), hi havia el cas d’una altra família afectada. Amb la seva unió i recerca posterior, descobriren que al territori balear, actualment, hi ha uns 30 menors afectats per la mateixa síndrome. La família d’Áurea i tres més han creat un petit grup de suport. Així, a més, com les edats dels nens són molt variades les famílies poden veure el progrés de la malaltia i com arribarà a ser la seva vida adulta.
Síndrome de Fabry
El naixement de Iago va transcórrer amb normalitat, però en complir els dos anys, els pares observaren que estava per sota dels percentils, tant en pes com en altura. A això, se li sumaren episodis de vòmits que normalment acabaven en ingressos hospitalaris. Amb aquesta simptomatologia li feren una sèrie de proves genètiques que confirmaren el diagnòstic: Síndrome de Fabry. Segons el portal Orphanet, dedicat a les malalties rares i als medicaments orfes, la malaltia de Fabry és un trastorn del metabolisme causat pel dèficit de l’enzim alfa-galactosidasa A, lligat a mutacions en el gen GLA que codifica l’enzim. En paraules de Cati, la mare de Iago, «és una malaltia de dipòsit, és a dir, el seu cos no produeix un enzim que té com a funció netejar tots els òrgans. A Iago, amb dos anys [ara en té quatre], ja no li funcionaven bé els ronyons». Les majors afectacions són al cor, als ronyons, la pell i l’estómac. A més, se sol tenir intolerància al fred o, com és el cas de Iago, a la calor.
«La majoria de malalties minoritàries no tenen un tractament específic»
Aquesta malaltia té una incidència anual d’1 cas per cada 80.000 nascuts, segons les dades d’Orphanet que també adverteixen que si es tenen en consideració variants d’aparició tardana, la prevalença passa a ser d’1 cas per 3.000 nascuts. La majoria de malalties minoritàries no tenen un tractament específic, però en el cas de la síndrome de Fabry hi ha una teràpia de reemplaçament enzimàtic. Es tracta d’un medicament d’administració intravenosa al llarg de sis hores que s’ha de subministrar cada dues setmanes a l’hospital.
A partir del cas de Iago descobriren que tota la família materna està afectada. El pare de Cati és portador de la malaltia, encara que amb 72 anys mai ha presentat símptomes. Cati i la seva germana també en són portadores, encara que tampoc han desenvolupat aquesta síndrome. El cosí de Iago sí que pateix la malaltia i segueix el mateix tractament, encara que com té 18 anys ja el pot ingerir a través d’una pastilla. A més, just quan li detectaren la malaltia a Iago, Cati estava embarassada del seu altre fill. Mael, que ara té dos anys, també ha donat positiu, però de moment no ha mostrat a penes símptomes, més enllà de la intolerància al fred.
Síndrome de Coffin-Siris
Clara és una nena de cinc anys. Quan al final de l’embaràs feren els mesuraments del fetus, ja varen veure alguna anomalia, però després d’una amniocentesi van dir als pares que no era res alarmant. Quan va néixer, Alejandra, la seva mare, narra que Clara no menjava perquè no era capaç d’agafar el pit ni el biberó. S’havia d’alimentar a través d’una sonda gàstrica i tampoc dormia. Les poques estones on aconseguia descansar, patia hipotèrmies. Li varen fer una anàlisi genètica i li detectaren la síndrome de Coffin-Siris amb una mutació de novo (és a dir, no l’ha heretada sinó que s’ha format en el seu organisme de manera aleatòria).
Segons Orphanet, és un trastorn clínic i genèticament heterogeni del que hi ha poc més de 150 casos diagnosticats. El més característic de la malaltia són el retard cognitiu o de desenvolupament, falange distal del dit petit del peu i trets facials diferencials que s’observen amb el pas del temps. També es pot donar talla baixa, dificultats en l’alimentació, microcefàlia, manifestacions oftalmològiques, anomalies cardíaques i cabell escàs, entre altres.
Alejandra explica que els dos primers anys de vida de Clara varen ser els més durs: «Hem hagut de treballar molt amb fisioteràpia perquè tenia un to de musculatura quasi inexistent. No podia caminar, el seu cos no era capaç d’aguantar el pes del seu propi cap». A partir del tercer any, la situació va millorar una mica i s’inicià a menjar textures, tipus purè. De totes maneres, Clara no té l’instint bàsic de l’alimentació: «Ella no agafarà un cobert per ella mateixa i se’l posarà a la boca perquè no té sensació de fam, fred o calor», assegura la mare.
«Tot i que amb la creació de les associacions s’ha aconseguit donar-li més visibilitat al tema, en la investigació encara queda molt per fer»
Aquesta síndrome tampoc té un tractament específic. Es tracta d’intentar millorar la seva qualitat de vida al màxim grau possible i veure fins on pot arribar cada pacient. De totes maneres, Alejandra ho viu de manera molt positiva malgrat els durs inicis: «Una doctora em va donar el condol només veure la xiqueta, assegurant que aviat la perdria», explica.
Clara està escolaritzada en un col·legi d’educació especial, una manera de donar unes hores lliures a la mare, que assegura que les cures han de ser directes, contínues i permanents. Quan li varen diagnosticar la malaltia a Clara, només hi havia un altre cas igual a tota Espanya. En aquell moment, Alejandra va decidir crear l’Associació espanyola de Coffin-Siris. Actualment, 42 famílies comparteixen les preocupacions i inquietuds davant l’avanç de la malaltia.
El cas de Neus
Neus és una adolescent de setze anys. Pateix tres trastorns de manera combinada, però la malaltia que més l’afecta és la hiperlisinèmia. Arran del seu cas, Catalina, la seva mare, va fundar l’any 2009, l’associació Abaimar (Associació Balear de Nins amb Malalties Rares).

En la imatge, la presidenta d’Abaimar, Catalina Cerdà (al centre), rep una donació per part de representants de l’Associació Cosipatchword / Foto: Abaimar
Catalina narra que Neus va venir al món «sense instint de succió». Per tant, des del primer dia ja es va haver d’alimentar a través d’una sonda gàstrica. Li diagnosticaren: hiperlisinèmia, un trastorn que es caracteritza per presentar nivells elevats de lisina en l’organisme. La base de la seva dieta són les fruites i les verdures, aliments que ajuden a mantenir les quantitats d’aquest aminoàcid a nivells normals. Però també li van diagnosticar altres dues malalties, hiperpipecolèmia i mitocondriopatia.
Neus està matriculada en un institut ordinari del seu poble i es forma en les anomenades Aules UEECO o aules inclusives. Són espais específics i adaptats destinats a alumnat amb diferents discapacitats perquè puguin compartir les seves experiències i portar una formació de manera normal i quotidiana. A més, aquests espais estan dotats per a respondre a totes les necessitats mèdiques que puguin presentar els afectats.
El paper de les associacions
La creació d’Abaimar va marcar un abans i un després al territori balear en el món de les malalties rares. La seva fundadora, Catalina Cerdà, juntament amb una altra mare en la mateixa situació, descriu que mai havia sentit parlar d’aquests trastorns: «Quan per loteria et toca a tu, tens moltes preguntes sense resposta. A més, després del diagnòstic, a l’hospital no vaig tenir ajuda de ningú. No sabia com gestionar les nostres vides, ni com portar la paperassa de la discapacitat i dependència…». Per tant, la voluntat d’aquestes dues mares afectades fou construir una comunitat de reforç i suport per poder ajudar a altres famílies en la mateixa situació.
Des de ja fa uns anys, Abaimar compta amb subvencions públiques, encara que també tenen donacions privades d’empreses o particulars. Amb aquestes ajudes i la quota dels socis, l’associació pot oferir als seus usuaris serveis: treballadors socials (per a tramitar documentació de discapacitat, dependència, ajudes, informes socials, etc.), equip de psicologia, musicoteràpia (ajuda a l’estimulació dels menors) i fisioteràpia. A més, també col·laboren amb una associació de la Universitat de les Illes Balears, IneDhitos, per a oferir reforç escolar domiciliari als seus socis.
En l’àmbit estatal, Abaimar està adscrita a FEDER que va nàixer el 1999 «amb l’objectiu de ser l’altaveu dels més de tres milions de persones que conviuen amb alguna d’aquestes patologies al nostre país». Actualment FEDER acull 370 entitats adscrites, el que representa a més de 1.300 patologies i quasi 97.000 persones. FEDER també treballa amb EURORDIS, associació d’àmbit, i ALIBER, una aliança iberoamericana.
Més investigació
Tots els testimonis de mares amb fills que pateixen una malaltia rara comparteixen una opinió unànime respecte aquest tema: falten molts anys d’investigació i d’estudi per a poder millorar la qualitat de vida dels pacients de malalties rares. Tot i que amb la creació de les associacions s’ha aconseguit donar-li més ressò i visibilitat al tema, en el camp de la investigació no ha seguit el mateix curs i encara queda molt per fer. Com explica Áurea, la informació que tenen disponible sobre la malaltia dels seus fills «és la mateixa de fa deu anys». També per a Alejandra, la investigació i la descoberta de tractaments pal·liatius són necessaris: «cal poder detectar la malaltia durant l’embaràs i saber com condicionarà la vida de l’infant». Tot un repte per a unes malalties que volen deixar de ser invisibles.